CRISPR-Cas9 Extracellular Vesicles for Treating Hearing Loss
Xiaoshu Pan , Peixin Huang ,Samantha S. Ali ,Tarun E Hutchinson
The treatment of inner ear disorders remains challenging due to the intrinsic anatomical barriers. The majority treatments and delivery approaches for accessing inner hair cells are still engaged with surgical intervention, which is highly invasive and inconsistent in terms of efficacy and safety. In order to address this challenge for crossing anatomical barriers, we report an extracellular vesicle (EVs) -based delivery approach to inner hair cells, which enables carrying CRISPR/Cas9 ribonucleoprotein (RNP)-sgRNA complex in high-throughput and high efficiency. The novel Microfluidic Droplet-based Electroporation System (µDES) is developed to efficiently load cargos into EVs via millisecond pulsed, low-voltage electroporation within flow-through droplets as enormous bioreactors in a continuous-flow and scalable manner. The observed loading efficiency of CRISPR/Cas9 RNA complex into EVs (RNP-EVs) is 10-fold higher than current bulk cuvette electroporation with hundred-fold increase of processing throughput. The low-voltage electroporation minimized the Joule heating influence on nanosized EVs, which retained the native surface membrane properties of cargo-loaded EVs. Both ex vivo and in vivo testing in Shaker-1 mice model demonstrated the high biocompatibility and biodistribution of produced RNP-EVs in the mouse cochlea penetrating inner hair cells. In contrast, the CRISPR/Cas9 RNP lipid-like nanoparticles (RNP-LNPs) control group was unable to penetrate anatomical barriers to access inner hair cells. In the Shaker-1 mouse model, DES produced RNP-EVs demonstrated much higher editing efficiency at Myo7ash1 mRNA level and showed significant hearing recovery in the Myo7aWT/Sh1 mice via Auditory Brainstem Response (ABR) testing. The report work will present a new solution to advance gene therapy in treating sensorineural hearing loss .
We kindly thank the researchers at University of Florida for this collaboration, and for sharing the results obtained with their system.
Introduction
Hearing loss is one of the most common neurodegenerative disorders with genetic causes in human affecting more than 450 million people worldwide1-2. In situ delivery of functional gene materials to cochlear hair cells is one of the most promising strategies to repair hair cells and restore hearing function in vivo1,3-6. To date, the gene therapy targeting cochlear hair cells are heavily relied on engineered AAV vectors that can transduce inner hair cells more efficiently. However, a few biosafety investigations of high dose AAV vectors in non-human primates are limiting the clinical translation5,7. On the other hand, the commonly used AAVs in gene therapy have limited capacity on cargo size (∼ ≤ 5Kb), which is unable to carry CRISPR SpCas9-gRNAs, as well as the Myo7a gene (∼100 Mb or a cDNA of ∼ 7 Kb) we studied in this work as a hearing loss causative gene in inner ear hair cells8. Although the lentiviruses have a cargo capacity of ∼10 Kb, the risk of insertional mutagenesis and severe immunogenicity are still significant concerns for clinical translation9. Alternatively, extracellular vesicle (EVs)-based delivery is emerging as a novel, safe approach for addressing such challenges employed in gene delivery10-13, owing to the intrinsic biocompatibility, low immunogenicity, tissue penetration ability, and superb tunability14-17. Although, EVs have been utilized to deliver various genes into tissues, the delivery of CRISPR/Cas9 ribonucleoprotein (RNP)-sgRNA complex has not been explored for inner ear tissue yet. The first-in-human trial using umbilical cord mesenchymal stromal cell derived EVs demonstrated their regenerative potential to attenuate inflammation-based side effects from cochlear implantation and noise trauma10,18, which indicates natural distribution of EVs across anatomical barriers in cochlear may present, making EVs more favorable in hearing loss gene therapy than its counterparts, viral vectors.
However, loading CRISPR RNP complex into EVs has been a grand challenge. Current methods suffer from low loading efficiency and not scalable. For instance, chemical transfection rate for CRISPR RNP complex is generally below < 25%, and the produced EVs are in low stability19. Utilizing cells engineered as the primary EV producer is limited with cargo type and copy numbers that can be passed to EVs for encapsulation. Although electro-transfection is more efficient in terms of transfection rate (∼ 50%), the scalability is limited with only a few milliliters of processing volume in their throughput20-21. Different from cells, EVs generally have much smaller size and higher Brownian motion. Therefore, we introduce a novel continuous-flow platform utilizing microfluidic droplet-based EV electroporation (µDES), which can handle variable cargos loaded into EVs in large throughput and high efficiency. The saturated cargo concentration in the confined uniform droplet bioreactors can maximize mass transport and electroporation efficiency. Such streamlined EV electro-transfection using continuous-flow droplets as the enormous micro-bioreactors has not been explored elsewhere. Compared to microfluidic nanoporation for EV transfection15,17,22-24, the continuous flow enables much larger scale and throughput processing (up to litter range). Only a low-voltage (∼10-30 volts) DC power is needed, which avoids Joule heating and thermal damage on nanosized EVs25. Compared to chemical transfection which introduces unpurifiable chemicals potentially toxic to in vivo system, the instant electric field application across flow-through droplets in millisecond (∼ms) minimizes perturbation of EV molecular components to retain the natural EV property and biocompatibility. We also employed FDA approved additive trehalose 26-30 in the buffer system to preserve EVs in good stability29, 31, and minimize membrane aggregation26-27 and leakage after electro-transfection as reported by other research28, which is suited in clinical settings.
In the realm of hearing loss, CRISPR/Cas9 technology are demonstrating promising editing specificity and efficiency by targeting and correcting genetic mutations responsible for various hereditary hearing disorders32. For instance, it can address mutations in TMC1Bth crucial for the development of sensory hair cells or cochlear function, potentially restoring auditory function33. More pioneer investigations exhibited that CRISPR/Cas9 technology can be applied to both congenital and acquired hearing loss, offering a multifaceted approach to treatment3,7,33-35. In this work, we target on Myo7a gene which plays an essential role in the development and maintenance of auditory hair cells. Myo7a mutation has been identified as the major causative gene (39–55% of the total cases) in Usher syndrome (USH1B), syndromic and non-syndromic hearing loss (DFNA11 and DFNB2), and age-related hearing loss36-37. Thus, timely removal of mutant myoVIIa allele could prevent progression of hearing loss. However, current Myo7a gene therapy is unattainable, due to limited options of vectors. Our work on both ex vivo and in vivo testing in Shaker-1 mouse model demonstrated the high biocompatibility and biodistribution in the inner ear tissue from our μDES produced RNP-EVs in mouse cochlea penetrating into inner hair cells. The RNP-EVs displayed much higher editing efficiency at Myo7ash1 mRNA level and showed significant hearing recovery in Myo7a WT/Sh1 mice via Auditory Brainstem Response (ABR) testing. In contrast, the CRISPR/Cas9 RNP lipid-like nanoparticles (RNP-LNPs) control group was unable to penetrate anatomical barriers to access inner hair cells. Our approach will allow the rapid loading of CRISPR into EVs for delivery of Cas9 without using a split vector, which offers the opportunity to customize sgRNAs addressing different mutant alleles within one gene, and enable customization to patient genetic heterogeneous mutation background, leading to a clinically translatable approach for overcoming current challenges in gene therapy.
RESULTS
High throughput and highly efficient EV electro-transfection via μDES platform
We developed µDES platform for enhancing EV transfection efficiency, loading capacity, and throughput. The concept of µDES platform and functionality are illustrated in Figure 1 A-E. The device composed one aqueous inlet with purified EVs and RNP cargos, one oil inlet, electroporation chamber and one droplet outlets which streamlines droplet generation with electroporation using a low voltage DC power supply. The device fabrication was detailed in supplemental materials. Continuous generation of droplets uniformly as enormous bioreactors in fast speed enables large-scale encapsulation of EVs with high concentration cargos (Figure 1F). In droplet space, the cargo transport under electric field is more efficient to cross transient pores from EV membrane via electrical mobility of cargo themselves, electric flux, and concentration gradient, which only needs milliseconds to complete in such small scale, in turn, maximizing the loading efficiency and capacity (Figure 1B). The uniform electric field distribution can be formed across each flow-through droplet for highly efficient electroporation as proved by COMSOL simulation in Figure 1C. Notwithstanding, droplet-based electroporation could also lower the dispersity of electric field in the small volume and contribute to more homogeneous electric distribution in small volume range38-40. The COMSOL simulation of μDES device was conducted which showed uniform distribution of both flow profile and electric field profile with focusing on the droplet passing through the electroporation chamber (Figure 1 D and E). The scalability of μDES was also studied by collecting droplets in a large container (Figure 1F) with uniform size of produced droplets (Figure 1G). By using fluorescently tagged 100nm polystyrene beads as the reference particles comparable in size to EVs, it showed that good encapsulation of fluorescent signal from beads in the droplets without any significant signal outside the droplets (Figure 1H). To perform the emulsion of water-in-oil, fluorinated oil FC40 and associated fluoroSurfactant was employed to generate oil phase due to their low conductivity, chemical inertness and stability, and easy removal. Pharmaceutical grade FC40 oil is considered as the highly biocompatible and low-cost recipe for droplet generation employed in pharmaceutics and in compliance with FDA 41-42. We also introduced the pharmaceutical grade trehalose as stabilization additive in the buffer during the electro-transfection, which can enhance the EV stability to minimize membrane fusion and leakage, in turn, improve the electro-transfection efficiency as documented in literatures26-30, 43. The droplet size which determines the throughput can be controlled by adjusting the pressure/flow rate of water-to-oil ratio44. For achieving high throughput, the droplets can be generated in high speed (∼700 droplets/min), which leads to ∼30 mL per hr processing throughput for each device. Note that current cuvette electroporation only handles ∼100 μL per device. The electroporation power did not alter the droplet size and quantity (Figure 1 I and J). For the efficient purification of cargo-loaded EVs from excessive Cas9 cargos, we employed Ni Sepharose high performance magnetic beads to selectively capture the His-tagged Cas9 proteins. The cargo loaded EVs in aqueous phase can be collected via centrifugation through phase separation to fully remove oil phase (Figure 1A cargo-loaded EV collection).
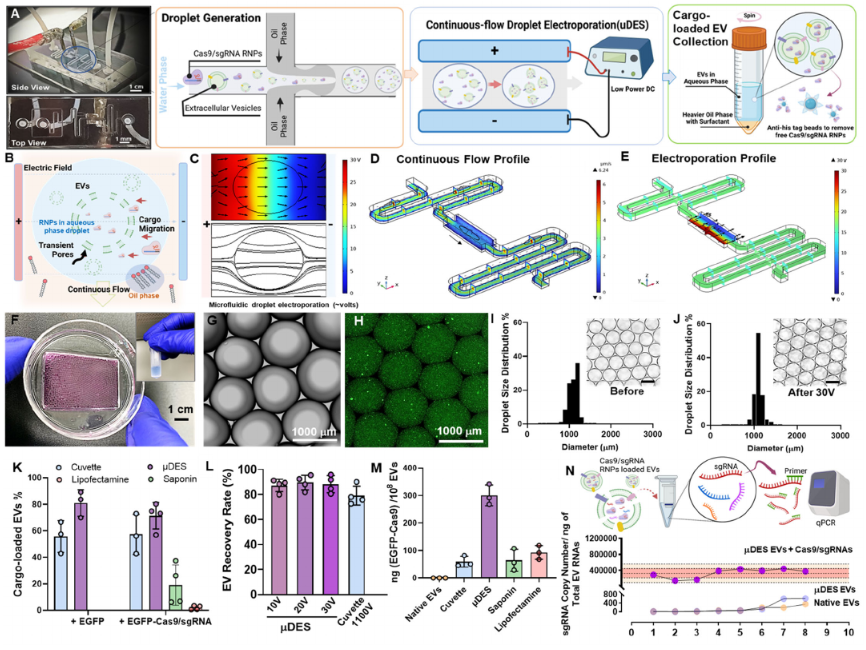
(A) Image of µDES device with illustration of continuous-flow droplet generation, droplet-based electroporation, and cargo-loaded EV harvesting and purification. (B) Schematic illustration of droplet-based electroporation of EVs under uniform electric field distribution as demonstrated by COMSOL simulation (C). The COMSOL simulation of continuous fluidic profile (D) and electric field profile (E) to show the uniformity for precision control. (F) Picture of large-scale collection of cargo loaded EVs in droplets. (G) Microscopic image of continuous flow generated droplets with green fluorescence stained nanobeads mimicking EVs encapsulated inside (H). The droplet size is uniform before(I) and after (J)electric field application for transfection (30 V). The insert scale bar is 1000 μm. (K) Evaluation of EV cargo loading rates among different transfection methods using fluorescence nanoparticle tracking analysis (FNTA). (L) EV recovery rate evaluated by NTA compared with conventional cuvette electro-transfection which requires 1100 volts for electroporation. (M) Quantitative measurement of CRISPR Cas9 proteins from transfected EVs normalized by EV particle number among different transfection methods. (N) The quantitative PCR analysis of transfected sgRNA copy number normalized by total EV RNAs. The electro-transfection was done by using μDES platform in 8 replicates. The native EVs and μDES prepared EVs without RNP cargo both served as the negative control groups. EVs are purified from HEI-OC1 ear hair cell culture.
Our platform can produce ∼80% transfection rate for large proteins including CRISPR Cas9/sgRNA RNP complex, which showed significantly higher efficiency than other conventional transfection methods including direct incubation, lipofection, and cuvette electro-transfection (Figure 1K). EGFP-Cas9 were pre-assembled with gRNA at 1: 2 molar ratio before the electroporation. EVs derived from HEI-OC1 cell, a putative progenitor hair cell line, were isolated and quantified for mixing with EGFP-CRISPR/Cas9 ribonucleoprotein (RNP) in electroporation low conductivity buffer. The final concentration of 1010 /mL of EVs was used for Neon cuvette electroporation as the control group, and our μDES system, as well as other chemical transfection methods. The findings revealed significantly enhanced cargo loading within EVs using μDES system, with a percentage as high as 80% for EGFP and 70% for EGFP-Cas9/sgRNA, surpassing cuvette electroporation and chemical transfection methods while maintaining minimal sample loss (Figure 1K). We also compared the recovery rate with conventional cuvette electro-transfection (Figure 1L), which showed better recovery due to continuous-flow harvesting. We quantified the RNP Cas9 protein loading amount (Figure 1M) to compare between different transfection methods, the μDES group exhibited more than 10-fold increase than other methods. The reproducibility of μDES loading was characterized using qPCR to quantify sgRNA loading amount from 8 replicates, with native EVs and µDES conditioned EVs without RNPs as control groups. Results demonstrated the good loading capacity and reproducibility, and no leakage or changes from intrinsic EV molecular components (Figure 1N). Overall, the μDES platform demonstrated the advanced performance on EV cargo loading.
Characterization of produced RNP EVs in high biocompatibility and tissue penetration
The µDES platform maintained a consistent flow rate, uniform electroporation pulse periods for each droplet and efficient cargo loading into EVs, therefore, retaining good EV properties as their natural un-treated EVs in terms of size (Figure 2A), zeta potential (Figure 2B), protein contents (Figure 2C), morphology and surface properties (Figure 2D). Results also indicated that μDES droplet oil phase with surfactant do not impose adverse influence on the produced EVs in aqueous phase due to phase separation. We tested the essential protein contents (CD81, TSG101, Alix) from µDES produced EVs, which is in line with native EVs in terms of expression level but carry significant amount of transfected Cas9 proteins (Figure 2C). The immune gold nanoparticle (AuNP) staining TEM imaging showed unnoticeable surface adsorption of Cas9/sgRNA RNPs from µDES produced EVs as compared with native EVs, which indicates RNA cargoes are loaded inside of EVs. By comparing with LNPs, we tested the cell biocompatibility (Figure 2E) and cellular uptake behavior (Figure 2F) via dosing HEI-OC1 ear hair cells with both µDES produced bone marrow mesenchymal stem cell derived EVs (RNP MSC-EVs) and HEI-OC1 hair cell derived EVs (RNP HEI-OC1 EVs). Both EV groups showed enhanced ability to promote ear hair cell growth compared with LNP group (LNP-102). The µDES produced RNP EVs did not show noticeable differences with their un-loaded native EVs. After one-hour cellular uptake, µDES produced EGFP-fused Cas9/sgRNA RNP MSC EVs exhibited higher uptake rate for cytoplasmic release and gradual entry into the nucleus (white arrow indication) compared with LNP group (Figure 2F). In order to further characterize the in vivo ear tissue biodistribution behavior, three groups of RNP*EGFP LNP, µDES produced RNP*EGFP MSC EVs and RNP*EGFP HEI-OC1 EVs were used via posterior semicircular canal injection into Shaker-1 mice ear individually. The confocal imaging from LNP group showed the ineffective distribution for entering into inner ear hair cells (Figure 2G and H). In contrast, both EV groups exhibited higher penetration into inner ear and uptake by both outer hair cells (OHCs) and inner hair cells (IHCs) (Figure 2 I and J). Thus, the results strongly support the feasibility of µDES transfected CRISPR RNP EVs employed in gene therapy delivering to inner ear.
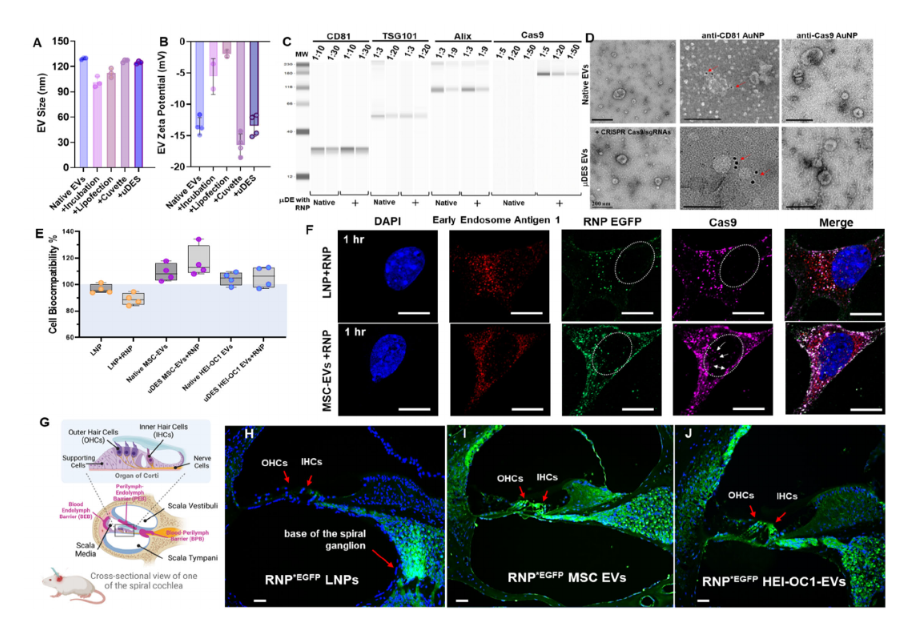
(A) Characterization of EV size and zeta potential (B) after transfection using different transfection methods with the original native EVs as the control group. (C) MicroWestern Blotting analysis of essential protein contents (CD81, TSG101, Alix, Cas9) derived from µDES produced RNP MSC EVs in serial dilution, with native EVs as the control group. (D) Immune gold nanoparticle (AuNP) staining TEM imaging analysis of µDES produced RNP MSC EVs with native EVs as the control group, in terms of CD81 surface marker expression and Cas9 surface identification. Scale bar is 200 nm. (E) Cell biocompatibility analysis using MTT assay with HEI-OC1 ear hair cells (∼106) dosed with LNP group (LNP-102, Cayman Chemical, w/o RNP), MSC-EV group (w/o RNP), and HEI-OC1 EV group (w/o RNP) in ∼109 particles. (F) Confocal imaging analysis of HEI-OC1 hair cell one-hour uptake dosed with RNP*EGFP LNP (LNP-102, Cayman Chemical) and RNP*EGFP MSC-EV group in ∼109 particles. The white arrow indicates the cytoplasmic release and gradual entry into the nucleus. Scale bar is 10 μm. (G) schematic illustration of the biological barriers in the structure of Corti and cochlea including blood endolymph barrier (BEB), perilymph endolymph barrier (PEB), and blood perilymph barrier (BPB). Confocal tissue imaging analysis of biodistribution in the organ of Corti via posterior semicircular canal injection of (H) RNP*EGFP LNP, (I) µDES produced RNP*EGFP MSC EVs and (J) RNP*EGFP HEI-OC1 EVs into Shaker-1 mice ears individually. Scale bar is 30 μm. All graphs show the mean ± SEM and biological replicates.


CRISPR design system for allele-specific editing of pathologic Myo7ash1
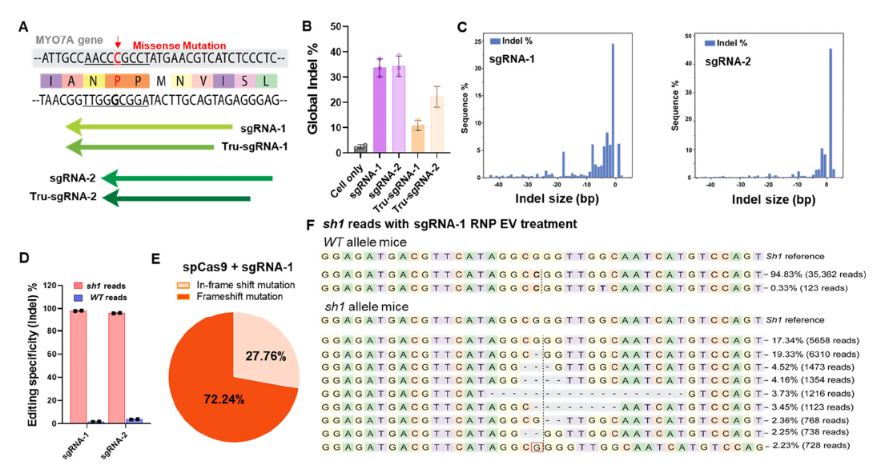
The Shaker-1 mouse model has been widely used for auditory research encoded by the deafness gene Myo7a, which is expressed very early in sensory hair cell development in the inner ear45. The Myo7a mutation makes up 4.5% of cases of sensorineural hearing loss evaluated in a large human patient cohort46, which presents significant clinical populations with a high level of burden that is not addressable by current therapeutic interventions. Timely removal of mutant myo7a allele could potentially prevent progression of hearing loss. Therefore, we demonstrated an allele-specific editing system using CRISPR/Cas9 with designed gRNAs targeting G-C mutations in hearing loss Shaker-1 mouse model. For in vitro validation using ear fibroblast cells from Myo7ash1/WT mice, we screened SpCas9 and two different gRNA sets harboring sh1 mutation modified with 2’-O-Methyl and 3’-phosphorothionate bonds on the last bases on 5’ and 3’ end (Figure 3A). In each gRNA set, we designed full-length and truncated forms targeting Myo7ash1 (supplementary Table 1). The gel electrophoretic analysis shows that CRISPR/Cas9 complexes efficiently cleaved targeted Myo7a amplicons (supplementary Figure 3). The T7 endonuclease assay was used to screen the allele-specific editing in vitro in Myo7AWT/WT, Myo7Ash1/WT and Myo7Ash1/sh1. The indel percentage is reduced to ∼25% in Myo7Ash1/WT when compared to ∼45% indel percentage in Myo7Ash1/sh1(supplementary Figure 4). The similar halving reduction in indel percentage is also observed in other gRNA designs when targeting Myo7ash1/WT. The data supports a robust editing selectivity when targeting Myo7Ash1/sh1 and Myo7Ash1/WT in vitro. Sanger and next generation sequencing was further used to confirm the allelic cleavage specificity and editing efficiency. ICE (Inference of CRISPR Edits, DeskTop Genetics) analysis of sanger sequencing data showed that a good amount of indel occurred only in Myo7ash1/WT, but not in Myo7AWT/WT(supplementary Figure 4). The sequence percentage analyzed by CRISPResso247 showed that gRNA-1 and gRNA-2 have higher cleaving activity than their truncated versions respectively (Figure 3B). The indel profile revealed that the majority of CRISPR-induced variants were deletions for gRNA-1 while insertions for gRNA-2 (Figure 3C). The sequencing variations demonstrated that 94.83% of Myo7aWT is unedited in Myo7ash1/WT. The small amount of substitution in 0.33% is possibly from PCR and sequencing background errors. In contrast, Myo7ash1 sequences is greatly reduced to ∼18% and the edited sequences account for the rest 82% in Myo7ash1 allele. We then quantified the targeting specificity of gRNA designs by sorting out mutation pattern 5’-CCG-3’ and wild type pattern 5’-CGG-3’ separately when targeting Myo7ash1/WT in vitro. By designing PAM sequence of gRNAs which have closer proximity to the Myo7a mutation, 95% specificity was achieved for in vitro editing (Figure 3D) and the wild-type allele is mostly intact after the CRISPR/Cas9 editing. Taken together, full-length of gRNA-1 and gRNA-2 have the best allele specific editing property in vitro thereby suggesting the potential for further in vivo test. Based on the global analysis of all the sequences in CRISPResso2 (Figure 3F and supplementary Figure 4), the most common mutation type is either single base deletion or single base insertion causing a frame shift in the coding sequence of Myo7a protein. In summary, SpCas9-gRNA-1 and SpCas9-gRNA-2 sets were specific for the Myo7ash1 allele and robust in interfering the shaker-1 mutation at DNA level, indicating the promise for in vivo investigation.
CRISPR loaded EVs for in vivo restoring the progression of the hearing loss
The Shaker-1 mouse wild type (+/+) and heterozygotes (+/-) initially both have normal hearing. However, the mutant heterozygotes (+/-) animals will gradually progress hearing loss till 6 months of age to be completely deaf, which serves as a good model for testing gene therapy in treating hearing loss in vivo. We found that myo7a point mutation could result in the excess oxidative stress in inner hair cells48-49 compared to wild type animals due to the functional damage of hair cells, leading to develop a facile and straightforward assay for effectively evaluating hearing ability based on oxidative stress markers. As demonstrated in Figure 4, we administrated µDES produced CRISPR RNP MSC-EVs (∼ 109 particles in ∼10 μL) via the posterior semicircular canal into the ear in heterozygote (+/-) mice, with the good hearing wild type (+/+) mice and non-treated heterozygote (+/-) mice as the control groups. After monitoring at the month 3 and month 6, the Shaker-1 organ of Corti tissues were extracted for immunohistochemistry based oxidative stress analysis. The oxidative stress markers 4-Hydroxynonenal (4-HNE) and 3-Nitrotyrosine (3-NT) were stained in green fluorescence. The heterozygous (+/-) mice show high expression level of both oxidative stress markers 4-HNE and 3-NT at 3 and 6 months of age in the inner ear hair cells (Figure 4 white arrows), in contrast to normal hearing wild type (+/+) mice with no oxidative stress marker expression. Interestingly, in our treatment group of heterozygous (+/-) mice, both 4-HNE and 3-NT expression level indicating the oxidative stress was significantly reduced to the unnoticeable level at 6 months of age, indicating the recovery of hair cell auditory function and elimination of pathogenic myosin VIIa allele.

Immunohistochemistry for oxidative stress in the Shaker-1 organ of Corti. The inner hair cell is indicated by the white arrow. Primary staining with 4-Hydroxynonenal (4-HNE) 1:50, and 3-Nitrotyrosine (3-NT) 1:50. Secondary staining 1:1000, nuclei labelled with DAPI. +/+, wild type; +/-, heterozygous Shaker 1 mice. Heterozygous mice show labelling for oxidative stress markers at 3 and 6 months of age whereas wild type mice show no labelling. Heterozygous mice treated with Crispr-EVs that eliminate the pathogenic myosin VIIa allele show no evidence of oxidative stress at 6 months of age, indicating the recovery of inner hair cell auditory function. Scale bar is 150μm.
To further evaluate the gene editing efficiency at molecular precision in vivo using our µDES produced CRISPR RNP MSC-EVs, we administrated EVs (∼ 109 particles in ∼10 μL) via the posterior semicircular canal into the left ear with right ear as the untreated control group in Shaker-1 heterozygotes (Figure 5A). The tissue of Corti was extracted in week 4 after injection for mRNA sequencing (Figure 5B-C). The heterozygous Shaker 1 mutant/wild type mice hearing ability after the treatment of RNP-EVs was followed over six months of age (Figure 5D-F) based on the hearing threshold results from ABR test. In first 4 weeks of treatment, the μDES produced CRISPR EVs group already exhibited the gene changes at the mRNA level, which showed improved gene editing ability compared with RNP-LNPs and cuvette transfected RNP-EVs (Figure 5B). Such gene editing performance differences could potentially be due to the ability and efficiency of delivery carriers entering into the hair cells. The editing efficiency was amplified at mRNA level given the fact that mRNA of Myo7a was only transcribed in hair cells, although myo7a gene exists in every cell type in the organ of Corti.

A) Schematic illustration of animal testing schedule. (B) The indel% mRNA sequencing analysis of Myo7a sequence changes from extracted Corti tissue from Shaker 1 heterozygotes mice in week 4 after CRISPR EV injection, with RNP-LNPs and Cuvette transfection method as control groups. (C) Distribution of resulted sequences treated by μDES produced CRISPR MSC EVs in week 4 from mRNA sequencing analysis. The most common edited sequences were shown. (D) qPCR analysis of Myo7a fold change from extracted Corti tissue in month 6 after injection of mDES produced CRISPR MSC EVs, which showed significant reduction of mutant Myo7a in treatment group. (E) Representative auditory brainstem responses (ABRs) recorded from shaker-1 heterozygotes mice left ear treated with μDES produced CRISPR MSC EVs in month 6, and right ear without treatment. The start showed the hearing ability baseline (F) The groups of shaker-1 heterozygotes mice in p30 as the normal hearing control group, and p120 developing severe hearing loss, to compare with p120 group treated with μDES produced CRISPR MSC EVs, which indicates positive therapeutic function for preventing progression of hearing loss.
At 6 months, the pathogenic myosin VII a allele was nearly removed (Figure 5D). These mice at P30 have normal hearing serving as the control group in Figure 5F blue line, and later on developed a severe sensorineural hearing loss by six months of age in Figure 5F red line Het P120. In contrast, the treated p120 group of mice displayed significant restoration of hearing ability (black line) compared to untreated p120 group of mice (red line). There is no significant difference from treated p120 group of mice (Black) with p30 normal hearing group of mice (Blue), indicating that our developed CRISPR EVs can remove pathogenic myosin VII a allele in these heterozygous mice for restoring hearing ability.
Discussion
The development of gene therapy correcting or eradicating genetic mutations for restoring functional protein expression is essential in maintaining sensory mechanotransduction and regenerating cochlear hair cells, thereby, to improve hearing function8. It not only requires highly targeted gene editing complexed, but also most effective and cell-specific delivery platforms. Presently, more challenges with the application of viral vectors or lipid nanoparticles were found to deliver functional genetic materials into auditory sensory system, due to the low tolerance from sensitive hair cells to toxicity and the intrinsic anatomical barriers. The safety of the treatment is concerned with the transgene and constitutive expression of gene editing complexes from eukaryotic organelles, as well as the ototoxicity from chemical compounds, for future clinical translation. Using EVs to encapsulate CRISPR/Cas9 gene editing agents provides a transient way to target auditory hair cells for achieving permanent genetic alteration on the deafness gene such as shaker-1 allele. Such treatment prevents supplementary treatment to attenuate the potential ototoxicity induced by delivery vehicles or overexpression of CRISPR/Cas9 complexes in vivo. More importantly, EVs exhibited excellent tissue penetration ability to specifically access and target inner hair cells as we observed in our shaker-1 mouse model (Figure 2).
Droplet-based electro-transfection has been reported to have higher mass and heat transfer demonstrated in single cell electroporation. EVs derived from donor cells also consist of lipid bilayer and similar membrane proteins composition on cells. However, nanosized EVs contain more compact membrane curvatures and strong brownie motion. Using microfluidic droplet-based electroporation to maximize the encapsulation of cargos into EVs, in turn, maximize the gene editing efficiency in vitro and in vivo, has been demonstrated with approximately 10-fold increase on the loading amount of CRISPR/Cas9 RNP into EVs. The throughput is hundred-fold increase compared with conventional cuvette electro-transfection method. Fast continuous flow through with droplet also prevents direct contact of EVs with electrodes for retaining EV natural integrity and stability. Such platform offers an easily amenable approach for scaling up by integrating multiple chip units for future GMP grade manufacturing of cargo-loaded EVs. The rapid loading of CRISPR RNP into EVs will allow the delivery of Cas9 without using a split vector approach, which enables customization of sgRNAs for addressing different mutant alleles within one gene, thus, opening a new avenue for personalized precision gene therapy via tailoring patient genetic heterogeneous mutation background.
Materials and Methods
Materials and reagents All chemical reagents and materials were purchased from ThermoFisher unless otherwise specified. Modified gRNAs were synthesized and quantified by Synthego. NLS-spCas9-NLS, EGFP-spCas9-NLS nucleases, anti-Cas9 antibody (Clone 4A1) were purchased from Genscript. All of DNA oligos were purchased from Integrated DNA Technologies. The collagenase IV was obtained from STEMCELL Technologies. SYLGARD™ 184 Silicone Elastomer Kit was purchased from Dow Silicones Corp. Master Mold for PDMS device was obtained from CADworks3D. Q5 high-fidelity DNA polymerase was purchased from New England Biolabs. Human bone marrow-derived conditioned culture medium and human umbilical cord-derived conditioned culture medium for extracellular extraction were purchased from EriVan Bio. SM-102 LNP in ethanol was the generous gift from Dr. Fan Zhang, in the College of Pharmacy at the University of Florida. 6nm Goat anti-Mouse and anti-Rabbit, IgG, Immuno Gold reagents were purchased from AURION.
Droplet generator on chip
The 3D structure of microfluidic device was designed and drawn by SOLIDWORK CAD. The resin mold for PDMS casting containing 200 µm flow and 500 µm electrode channels, 150 µm as nozzle for droplet generation, was printed by µMicrofluidics Printer (CADworks3D, 30 µm resolution). Briefly, CAD files was opened by utility.exe that is connected to µMicrofluidics Printer. The software setting for printing as follows: 50 µm as thickness, 0.1 for grid size, 40% Power ratio. The microstructure was then sliced and ready to be launched for printing. The resulting resin mold was then soaked in 100% Ethanol or isopropanol for 10 min to remove the free resin before the final UV curing step. And then the resin mold was dried with compressed Nitrogen. The soaking-drying cycles have to be repeated several times until there is no shiny free resin on the microstructure. Each side of resin mold was then cured in the Creative Cure Zone (CADworks3D) for another 10min twice for further photopolymerization and solidification of microstructures. The resin mold was then ready for PDMS casting. PDMS was prepared using the standard 10 : 1 (base to curing agent) ratio. The PDMS mixture was stirred completely at least 3 min and then degassed for at least 30 min before being poured into the 3D-printed molds and then baked at 75°C for 3 hours. After the surface activation of molded PDMS pieces using a corona discharger and the microscope plate, PDMS molds were then assembled and bound onto the microscopic plate as the droplet-based electro-transfection device.
Electrodes with L shape were tailored to fit into electroporation chambers designed in μDES and then manually inserted into the electroporation sites to align well with each other. The 1/16 OD, 1/32 ID tubings were then inserted into the inlets and outlets in μDES. To avoid any potential pressure leakage that can result in the unstable flow rate in μDES, the additional PDMS was then added to the area surrounding the inlets, outlets and electroporation sites before use. And then the device was baked in the oven at 75°C for 30min. The resulting μDES is ready to the following electroporation.
COMSOL Simulation
The proposed microfluidic device was fine-tuned using the COMSOL Multiphysics software package. The device’s mathematical model involves fluid flow and electromagnetism. For both models a standard linear triangular extra-fine mesh was assigned to the geometry. To observe the geometric evolution of our droplets, we used the computational fluid dynamics system (CFD) module using the laminar two-phase flow. For this simulation, the oil phase material was defined as FC-40 with a density of 1850 kg/m3 and a dynamic viscosity of 0.0018 Pa/s. Microfluidic flows are defined by the Navier Stokes equation where ρ is the density of the fluid, u is the velocity of the field, t is time and P is the pressure field:
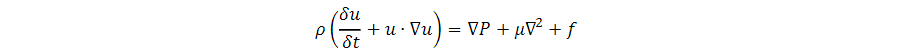
The physics of the electroporation system of the device were defined using the AC/DC module to simulate the electric field distribution in the microfluidics electroporation model. The material of the droplet was defined as electrolytic buffer with a conductivity of 1×10-4 S.m-1. The electrical conductivity of the electrode was defined by the composition of the Platinum-iridium wire as 9.43×106 S.m-1. In steady conditions, the flow of electric currents within a conducting fluid follows Ohm’s law:
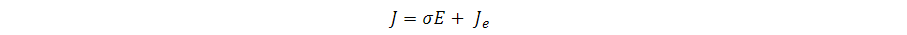
Here, J represents the total current density within the material while σ represents the electrical conductivity measured in S.m-1. E is the electrical field strength measured in V.m-1 and Je represents the current density. Moreover, to mathematically describe the electric field that acts upon the droplet within the microfluidic device, we use the induced potential difference ΔΨi at a point M of the droplet membrane at any time t
