Microfabricated dynamic brain organoid cocultures to assess the effects of surface geometry on assembloid formation
by Camille Cassel de Camps, Sabra Rostami, Vanessa Xu, Chen Li, Paula Lépine, Thomas M. Durcan and Christopher Moraes
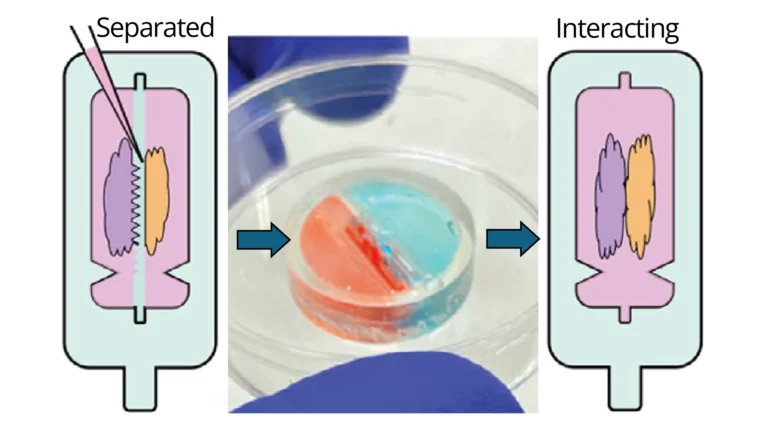
Abstract: Organoids have emerged as valuable tools for the study of development and disease. Assembloids are formed by integrating multiple organoid types to create more complex models. However, the process by which organoids integrate to form assembloids remains unclear and may play an important role in the resulting organoid structure. Here, a microfluidic platform is developed that allows separate culture of distinct organoid types and provides the capacity to partially control the geometry of the resulting organoid surfaces. Removal of a microfabricated barrier then allows the shaped and positioned organoids to interact and form an assembloid. When midbrain and unguided brain organoids were allowed to assemble with a defined spacing between them, axonal projections from midbrain organoids and cell migration out of unguided organoids were observed and quantitatively measured as the two types of organoids fused together. Axonal projection directions were statistically biased toward other midbrain organoids, and unguided organoid surface geometry was found to affect cell invasion. This platform provides a tool to observe cellular interactions between organoid surfaces that are spaced apart in a controlled manner, and may ultimately have value in exploring neuronal migration, axon targeting, and assembloid formation mechanisms.
We kindly thank the researchers at the McGill University for this collaboration, and for sharing the results obtained with their system.
1. Introduction
Organoids have gained popularity as experimental models for developmental and disease studies.[1-11] Grown from stem cells, these 3D tissue-engineered cultures can differentiate toward diverse lineages that capture the complexity of in vivo tissues.[1-9] Multiple organoid types can also be assembled to interact, fuse, and mature[12-14] and these “assembloids” can hence capture some of the additional cellular diversity and architectural complexity of multi-component organ systems, compared to single-type organoids.[12, 13, 15, 16] In the developing brain, for example, complex circuits are established by neuronal projection and migration to create both local and long-distance connections.[13, 17-19] Regionalized organoids can hence be assembled to create in vitro models of the circuits that run throughout the brain. For example, functional synaptic connections can form between cortical and striatal organoids, specific neurons can migrate from ventral to dorsal forebrain organoids,[20, 21] and muscle contraction can be stimulated by brain organoid activity.[22] Assembloids can hence be powerful in vitro models for a wide variety of neurodevelopmental disease processes.[20, 21]
Tissue geometry is now well-established to influence fundamental cellular processes, such as proliferation, differentiation, branching, and invasion.[23-30] Driven by endogenous mechanical cellular stresses that spontaneously arise in three-dimensional tissues,[24, 25, 31] these cellular phenotypes drive feedback loops that govern tissue organization, specification, and maturation.[23, 26, 32] While previous studies have demonstrated that geometric confinement and associated mechanical stresses drive the organization of developing neural structures,[33, 34] whether these geometric features play a role in neural organoid development and assembloid formation remains an open question. Such experiments would require the technical capacity to simultaneously impose a specific geometry on independently cultured organoids, and control their relative positions before allowing them to interact. Moreover, such experiments would require long-term culture in biologically permissive and optically addressable formats. Given the intrinsic challenges associated with precisely manipulating soft living matter, technical innovations are required to better understand assembloid formation.
Recent developments in organoid culture models suggest a path to achieve these goals. Park et al. recently developed a microfabricated culture approach in which oxygen-permeable silicone inserts are used to restrict the size and shape of intestinal organoids as they grow into a hydrogel matrix.[35] This approach was successfully used to allow stem cell proliferation and maturation, while controlling the global geometry of mature intestinal organoids, such that diffusive transport of oxygen, nutrients, and waste was sufficient to prevent the formation of a necrotic core that commonly arises in large, dense tissues.[35] Although very promising, this strategy has yet to be demonstrated for other types of organoids. Inspired by this approach, here we develop a strategy to separately culture distinct brain organoid types in adjacent compartments, while shaping the surface geometry of the tissues; and explore this concept using two types of brain organoids. After establishing mature organoids, an insert separating the organoid compartments can be manually removed and replaced with a thin layer of extracellular matrix, allowing the precisely positioned organoids to begin forming an assembloid (Figure 1). To prove this concept, here we use various channel geometries to shape unguided and midbrain organoids. We demonstrate simultaneous axonal projections emanating from the midbrain organoids, and surface geometry-specific cell migration from unguided organoids. We hence propose that this technical innovation allows systematic investigation of the role of interacting surface geometries in assembloid formation.
2. Methods
Unless otherwise stated, all cell culture materials and supplies were purchased from Fisher Scientific (Ottawa, ON) and chemicals were from Sigma-Aldrich (Oakville, ON). The use of induced pluripotent stem cells (iPSCs) in this research was approved by the McGill University Health Centre Research Ethics Board (DURCAN_IPSC/2019-5374).
2.1. Device fabrication process
Molds were designed in Fusion 360 (AutoDesk), and printed on a ProFluidics 285D 3D resin printer using Master Mold Resin (CADworks3D) with a layer thickness of 50 µm. After washing with isopropanol, mold pieces were cured in a 36 W ultraviolet (UV) chamber overnight. Molds were designed for assembly into chambers with patterned features on both the base and lid (Figure 2A). Polydimethylsiloxane (PDMS) prepolymer and curing agent were mixed at a ratio of 10:1 w/w, poured into the chamber, and degassed under vacuum. The molded lid was then lowered slowly from one side to avoid trapping air bubbles in the chamber. The lid was then pressed down to displace excess PDMS. Tongue-and-groove convex/concave features in the chamber base and lid contained the PDMS prepolymer after chamber assembly. PDMS was cured overnight in an oven at 40°C to minimize shrinkage[36] and de-molded using 70% ethanol to help release the devices from the 3D printed resin.


2.2. Device preparation for organoid culture
Base devices were coated with dopamine to improve adhesion to Matrigel.[37] Briefly, dopamine hydrochloride was dissolved in 10 mM Tris buffer (pH 8.5; 2 mg mL−1), pipetted into the devices, and incubated overnight at room temperature. After treatment, devices were rinsed in reverse osmosis water, and dried with a stream of dry compressed air. To facilitate release from the devices and reduce adhesion to tissue cultures, the removable inserts were passivated with Pluronic® F-68 (5% in water) overnight at room temperature.[38, 39] Treated devices were rinsed in water, and dried with compressed air. All components were sterilized for 20 min in a 36 W UV chamber before assembly. Devices were assembled on a coverslip, which formed the bottom of the media reservoirs and allowed the device to be easily manipulated as a unit. For experiments without a removable insert, glass coverslips were used as the base surface, after functionalization with dopamine. Assembled devices were sterilized by UV for 45 min.
2.3. Cell culture
The AIW002-2 iPSC line (male) was used to generate unguided (previously referred to as “cerebral” organoids), and the TD22 iPSC line (male) was used to generate midbrain organoids, following established and characterized protocols[40, 41] with some previously-described modifications.[42] Both lines were obtained from The Neuro’s C-BIG repository and had undergone multistep quality control.[43] T47D human breast cancer cells (ATCC HTB-133) were used for preliminary validation experiments with the microfluidic device.
All cell cultures were maintained at 37°C with 5% CO2. T47D cells were cultured in Dulbecco’s Modified Eagle Medium with 10% fetal bovine serum and 1% antibiotic–antimycotic (complete DMEM). Media was changed every 3–4 days, and cells were passaged using 0.25% trypsin–EDTA at 80% confluence in a 1:5 dilution. The iPSC lines TD22 and AIW002-02 were maintained on Matrigel-coated plates in mTeSR1 complete kit media (Basal medium with supplement; STEMCELL Technologies, Cat No. 85850) with daily media changes. iPSC plated petri dishes were checked every day for spontaneous differentiation using a brightfield microscope. In cases where cells with morphologies different from iPSC colonies were detected, locations were marked and then in the BSC, cells on the marked locations were scraped off using a P200 pipette tip. Then, the media was collected, and the plate was washed with DMEM-F12 media supplemented with 1% antibiotic-antimycotic to ensure the removal of the scraped cells from the culture. iPSC cells were passaged with Gentle Cell Dissociation Reagent (StemCell Technologies) at 70% confluence. A ratio of 1:10 was maintained throughout all passages in which the colony pellet was broken down in such a way that each fragment contained between 10 and 15 cells. The homogeneity of the colony sizes in the sub-culture was assessed the next day by imaging with a brightfield microscope. At this step, colonies that were either too large or too small were scraped off and removed from the culture using the same method mentioned for removal of spontaneous differentiation.
2.4. Organoid culture
When iPSC cultures reached 70% confluence, cells were detached with Accutase (Gibco), resuspended in the appropriate media, seeded at 10,000 cells per well in 96-well round-bottom ultralow attachment plates (Corning Costar), and centrifuged for 10 min at 1200 rpm to aggregate the cells. Organoids were seeded so that they would be ready for Matrigel embedding simultaneously (Day 7 for midbrain, and Day 12 for unguided).
Media was changed every other day according to published protocols.[40, 41] For unguided organoids: human embryonic stem cell (hES) media (low basic fibroblast growth factor (bFGF), with ROCK inhibitor) was used on Day 0 (consisting of 400 mL DMEM-F12 + Glutamax, 100 mL Knockout Serum Replacement, 15 mL hESC-quality FBS (Gibco), 5 mL MEM- non-essential amino acids, 3.5 µL 2-mercaptoethanol, bFGF at 4 ng mL−1 final concentration, and ROCK inhibitor at 50 µM final concentration); hES media (low bFGF, no ROCK inhibitor) on Day 2; hES media (no bFGF, no ROCK inhibitor) on Day 4; neural induction media on Day 7 and 9 (consisting of DMEM-F12 + Glutamax, 1% N2 supplement, 1% MEM-non-essential amino acids (MEM-NEAA), and heparin at 1 µg mL−1 final concentration); cerebral organoid differentiation media without vitamin A on Day 12 and 14 (consisting of 125 mL DMEM-F12 + Glutamax, 125 mL Neurobasal, 1.25 mL N2 supplement, 62.5 µL insulin, 1.25 mL MEM-NEAA, 2.5 mL penicillin-streptomycin, 1.75 µL of 1/10 2-mercaptoethanol dilution in neurobasal, and 2.5 mL B27 supplement without vitamin A); cerebral organoid differentiation media with vitamin A on Day 16 onwards (made using B27 supplement with vitamin A).
For midbrain organoids: neuronal induction media was used on Day 0 (consisting of 25 mL DMEM-F12 + Glutamax + 1% antibiotic-antimycotic, 25 mL neurobasal, 500 µL N2 supplement, 1 mL B27 without vitamin A, 500 mL MEM-NEAA, 1.75 µL of 1/10 2-mercaptoethanol dilution in neurobasal, heparin at 1 µg mL−1 final concentration, SB431542 at 10 µM final concentration, noggin at 200 ng mL−1 final concentration, CHIR99021 at 0.8 µM final concentration, and ROCK inhibitor at 10 µM final concentration); neuronal induction media without ROCK inhibitor was used on Day 2; midbrain patterning media was used on Day 4 (consisting of neuronal induction media without ROCK inhibitor with the addition of Sonic Hedgehog (SHH) at 100 ng mL−1 final concentration, and Fibroblast Growth Factor 8 (FGF8) at 100 ng mL−1 final concentration); tissue induction media was used on Day 7 (consisting of 50 mL neurobasal, 500 µL N2 supplement, 1 mL B27 without vitamin A, 500 mL MEM-NEAA, 1.75 µL of 1/10 2-mercaptoethanol dilution in neurobasal, 12.5 µL insulin, laminin at 200 ng mL−1 final concentration, SHH at 100 ng mL−1 final concentration, FGF8 at 100 ng mL−1 final concentration, and 50 µL penicillin-streptomycin); final differentiation media was used on Day 8 onwards (consisting of 50 mL neurobasal, 500 µL N2 supplement, 1 mL B27 without vitamin A, 500 mL MEM-NEAA, 1.75 µL of 1/10 2-mercaptoethanol dilution in neurobasal, brain-derived neurotrophic factor (BDNF) at 10 ng mL−1 final concentration, glial cell line-derived neurotrophic factor (GDNF) at 10 ng mL−1 final concentration, ascorbic acid at 100 µM final concentration, db-cAMP at 125 µM final concentration, and 50 µL penicillin-streptomycin).
2.5. Device loading
Cell cultures were either loaded as single cells or as pre-formed organoids into the devices. Single cell cultures were obtained by trypsinization (T-47D, breast cancer line) or detachment (TD22 iPSCs, using Accutase as previously described[42]) and resuspended in undiluted Matrigel (Corning 356230) at concentrations of 8 × 106 cells mL−1 for T-47D, or 1 × 106, 3 × 106, or 1 × 107 cells mL−1 for iPSCs. All pipetting steps were performed with chilled pipette tips to prevent premature polymerization of the Matrigel. To load the organoids into the devices, they were pipetted directly into the loading ports in media with cut P200 pipette tips on Day 12 or 13 after seeding cerebral organoids, and on Day 7 or 8 for midbrain organoids. Once in the chamber, they were too big to pass through the channel restriction. Media was aspirated, leaving the organoids in the device, and replaced with undiluted Matrigel. All devices were incubated for 20 min at 37°C to polymerize the Matrigel. The appropriate media was added after polymerization, and replaced every 2 days.[40]
2.6. Insert removal
Once the organoids had grown and adopted the shapes defined by the compartment dimensions, the inserts separating the compartments were removed. One pair of tweezers was used to hold the base device down, while another was used to slowly pull the insert away. Media was gently aspirated from between the organoids, and replaced with Matrigel to fill in the space. Devices were left at room temperature for 5 min to allow the newly added liquid Matrigel to seep into the existing Matrigel, and then incubated for 20 min at 37°C. Final differentiation medium from the midbrain protocol,[40] with the addition of insulin at a concentration of 0.25 µL mL−1, was added to the well for this stage of combined culture. This media formulation was selected based on consultation and comparison of existing organoid and assembloid protocols and the function of each component,[21, 41, 44] and would need to be adjusted if other types of organoids were grown in the coculture device.
2.7. Tissue staining
Live CellTracker Green or Red were loaded into cells at 20 µM in media overnight at 37°C. Live/dead viability assays were performed with calcein AM and ethidium homodimer-1 (Life Technologies), diluted in media to 4 µM each and incubated with devices for 30 min at 37°C.
For immunostaining, Matrigel was first dissolved using Cell Recovery Solution (Corning; at 4°C for 20 min, twice). Devices were washed twice with phosphate buffered saline (PBS), fixed in 4% paraformaldehyde for 1 h at room temperature, and washed three times for 15 min each with PBS before storage at 4°C until staining. Whole mount staining was performed on organoids directly in the devices, using standard protocols.[42] Briefly, organoids were incubated in blocking buffer (0.5% Triton X-100 + 5% donkey serum in PBS) for 5 h at room temperature, then with primary antibodies diluted in blocking buffer overnight at 4°C. Organoids were then washed with PBS, three times for 15 min each, and then incubated with secondary antibodies and Hoechst diluted in blocking buffer overnight at 4°C. Organoids were washed again as before. Antibodies and stains were used as follows: anti-tyrosine hydroxylase (TH) at 1:200 (rabbit polyclonal, Pel Freez P40101-150), anti-β-tubulin III (Tuj1) at 1:300 (chicken polyclonal, Millipore Sigma AB9354), anti-tau-1 clone PC1C6 at 1:200 (mouse monoclonal, Millipore Sigma MAB3420), anti-glial fibrillary acidic protein (GFAP) at 1:250 (rabbit polyclonal, Millipore Sigma AB5804), anti-microtubule-associated protein 2 (MAP2) at 1:400 (chicken polyclonal, EnCor Biotechnology CPCA-MAP2), goat anti-chicken IgY H&L (DyLight® 488) at 1:500 (Abcam ab96947), donkey anti-rabbit IgG H&L (DyLight® 488) at 1:500 (Abcam ab96891), donkey anti-mouse IgG H&L (DyLight® 594) at 1:500 (Abcam ab96877), donkey anti-Rabbit IgG (H+L) Alexa Fluor™ 594 at 1:500 (Invitrogen A-21207), and Hoescht 33342 at 1:5000 (Invitrogen H3570). Immunostains were performed with a negative control (staining without primary antibody) to confirm that under these imaging conditions, any detected signals were not the result of non-specific binding or autofluorescence.
2.8. Microscopy and image analysis
Devices were imaged using an EVOS transmitted light microscope (XL Core) or an EVOS M7000 fluorescent Imaging System. Images were processed and analyzed in Fiji (NIH).[45] Pairwise stitching was performed using the Stitching plugin[46] when needed. Axonal projections were measured from the organoid surface to projection tip to obtain their length and angle. Cell migration distances were measured from the organoid surface to the edge of the migrating cell front, 2–3 days after removal of the separating wall.
2.9. Statistical analysis
Analyses were performed in R statistical software.[47] All data was confirmed to be normally distributed, with equal variances. The measurements of axonal projection lengths toward nearby organoids were normalized by lengths not directed towards a nearby organoid within samples, and then a two-sample, two-tailed t-test was used to compare between those axons that were directed towards midbrain organoids against those directed toward unguided organoids. The measurements of axonal projection angle for each organoid were used to calculate kurtosis, after centering distributions around the angle defined as toward the nearby organoid; one-sample t-tests were used to compare against random chance (i.e., uniform distribution, kurtosis of 1.8), and a two-sample, two-tailed t-test was used to compare between groups. One-way ANOVA was performed with Tukey post hoc comparisons for measurements of cell migration distance. All analyses for significance were carried out with α = 0.05.
3. Results
3.1. Device design for separated adjacent co-cultures
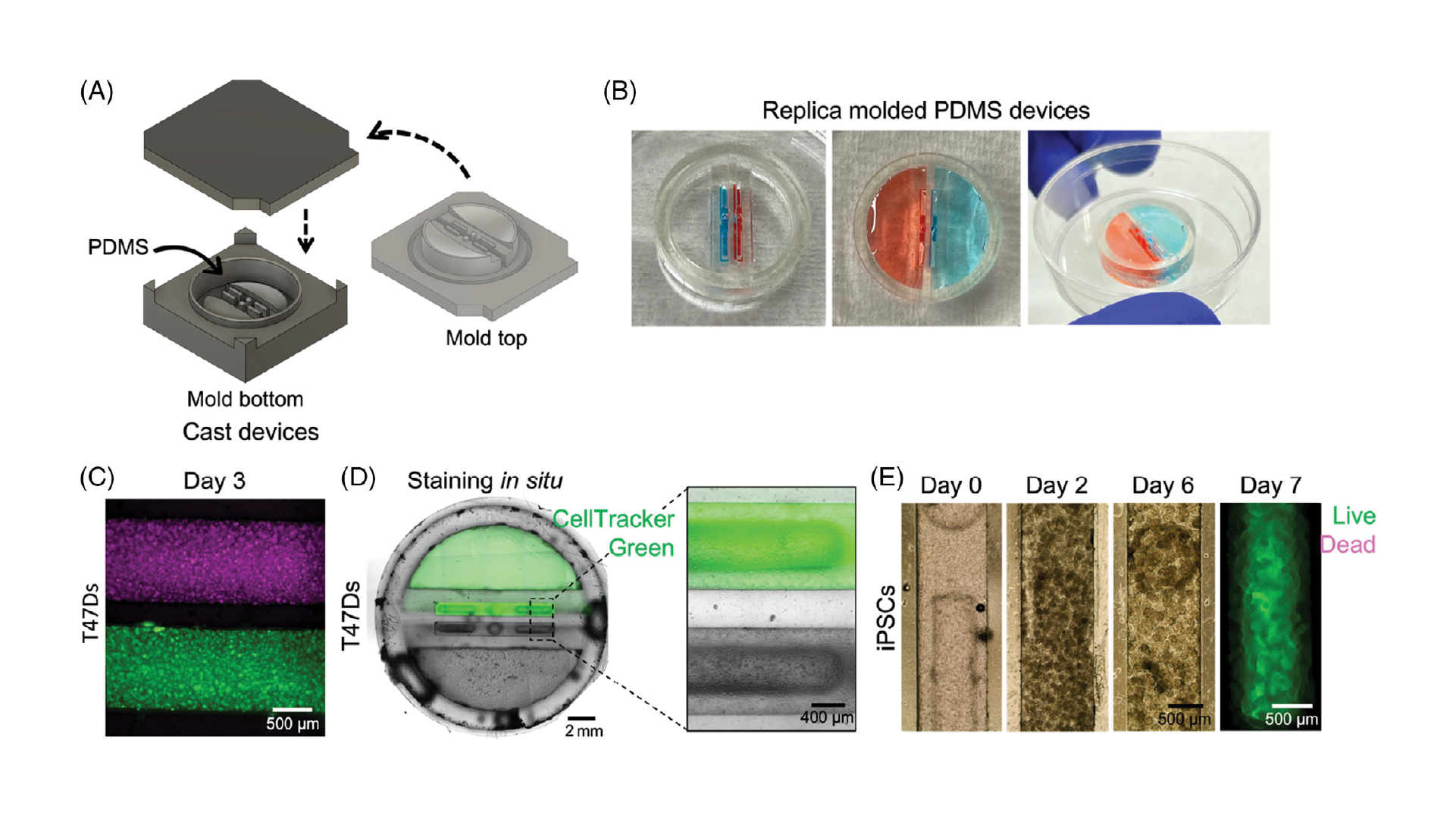
Double-sided 3D-printed molding chambers (Figure 2A) were essential for the successful operation of these devices, as complex 3D geometries and multiple overhanging and double-sided layer features were required, which could only be achieved by designing interlocking surfaces for double-sided PDMS molding. The PDMS devices themselves were designed to facilitate pipetting of Matrigel and cell/organoid solutions into the channels via inlet ports, while leaving the tops of the channels open for nutrient exchange. This was achieved using an overhanging phase guide that allows surface tension to hold injected liquid (prepolymerized Matrigel) in a confined space, while leaving a slit in the top of the channels open for media exchange (slit was 600 µm across). We adapted this design to create two adjacent channels, each fed by an integrated and independent media reservoir to support growth of organoids with separate media requirements (Figure 2B; shown with red and blue liquids to represent different media formulations).
We first verified that our devices were operational, suitable for cell culture, and that the media reservoirs were functionally isolated from each other by loading an available cell line (T47D breast cancer cells, used as a model cell line for preliminary experiments). We verified that the devices successfully separated cell compartments by dying the T47D breast cancer cells with either CellTracker Red or Green, and loading them in Matrigel into adjacent channels (each 1 mm wide, separated by ≈500 µm; Figure 2C). Devices were cultured for 3 days, and no color exchange was observed between compartments. Next, we sought to validate media reservoir function. T47D cells were suspended in Matrigel and loaded into channels. One reservoir was filled with regular media, and the other was filled with media containing CellTracker Green. Over several days in culture, only the cells in the channel fed by that reservoir were dyed green, demonstrating functional separation of the reservoirs produced by this fabrication technique (Figure 2D).
3.2. Devices support iPSC culture
Once device design was validated, we next sought to verify that the devices could support iPSC culture, which is typically much more stringent and would be required to grow developing organoids within the compartments. iPSCs were suspended in Matrigel at several different densities, loaded into the device channels as described, and cultured with midbrain organoid media.[40] Initially loaded at 10 million cells mL−1, the density of cells in the channels increased over days in culture, and multiple cells aggregated together to form numerous small clusters within the channels (Figure 2E). At 1 and 3 million cells mL−1, cells also aggregated to form extremely small clusters that grew over time (data not shown). We also confirmed that iPSCs were viable for at least 1 week in culture (Figure 2E) before proceeding with organoid culture experiments.
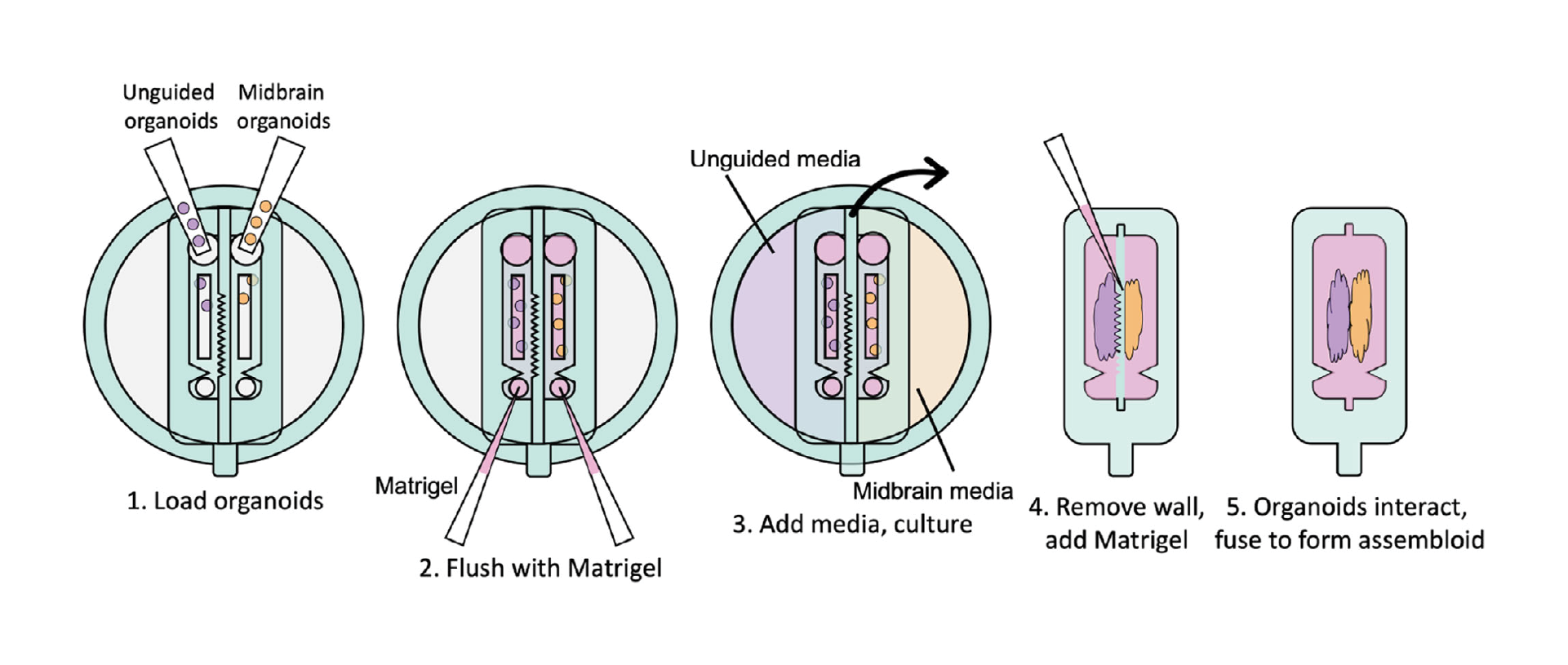
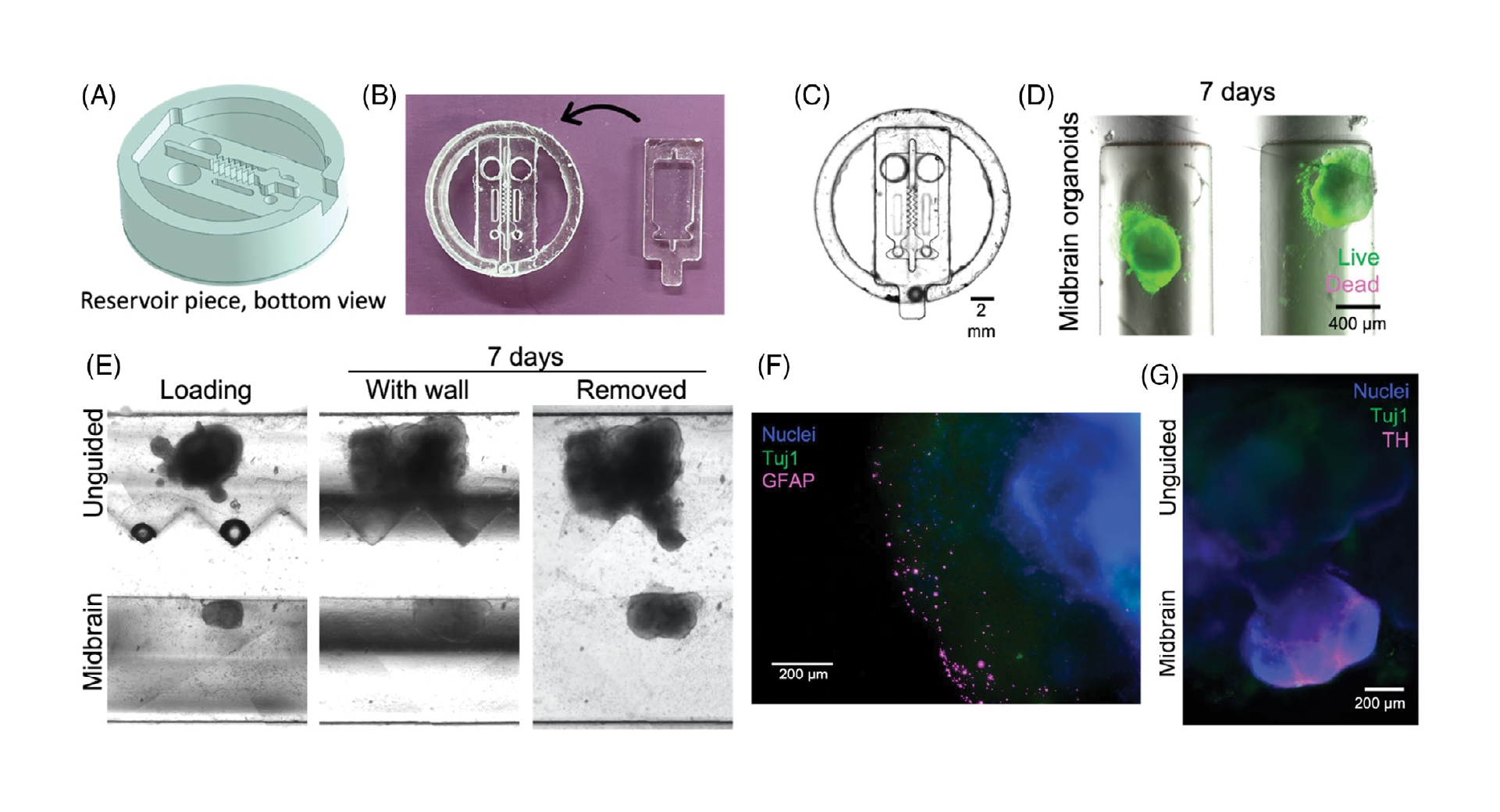
3.3. Removable inserts for dynamic organoid co-culture
Devices for dynamic organoid co-culture were fabricated as two separate pieces, the lower of which acts as a base to hold the organoids, while the upper piece includes the separating wall and media reservoirs. The separating wall can be designed with a variety of geometries to shape the growing organoids. The organoid loading ports were designed to be sufficiently large to load pre-formed organoids, as required in standard brain organoid culture protocols (Figure 3A–C), while a small outlet port was designed to allow Matrigel loading, while keeping the organoids in the channels. We then confirmed that midbrain organoids remained viable for at least one week in culture after loading (Figure 3D).
3.4. Devices support assembloid formation
As a first proof-of-concept, midbrain and unguided organoids were loaded in Matrigel in adjacent channels to form assembloids from two distinct organoid types. After 5–28 days, when the organoids had expanded to contact and mold themselves against the channel wall, the inserts were removed from the bases, leaving the organoids and surrounding Matrigel separated by the width of the separating wall (600 µm, but could be varied by mold design). This gap was back-filled with Matrigel, and the assembloids were then monitored during growth. We first confirmed that using this system, the organoids retained the shape of the insert wall after removal (Figure 3E). Immunostaining of fixed samples at this stage demonstrated that glial fibrillary acidic protein (GFAP)-positive astrocytes arise in unguided organoids only (Figure 3F), and tyrosine hydroxylase (TH)-positive dopaminergic neurons in the midbrain organoids only (Figure 3G), as expected for this type of organoid.[40] Within 3 days, the organoids bridged the space between them to initiate formation of an assembloid. These results confirm (1) appropriate and expected differentiation of these neural organoids within our devices, including differentiation towards the non-neuronal lineages expected to arise in unguided organoids[9]; (2) that our separated device supports distinct media-driven differentiation patterns in adjacent compartments; and (3) that assembloids can form after removal of the separating wall.
3.5. Axonal projections arise from midbrain organoids during assembloid formation
Having confirmed that the device architecture enables us to reliably examine a reproducible interface between organoids during assembloid formation, we then characterized the early stages of assembloid integration in terms of behavior of cells from each of the two organoids. We noted long and thin cellular processes arising from midbrain organoids, that began to appear after ≈7–9 days of co-culture in our devices (≈15–16 days after organoid seeding; Figure 4A). We confirmed that these were axonal projections by immunostaining for tau-1, which localizes to axons only,[48, 49] as well as for TH, which can also be found in axons[50] (Figure 4B).
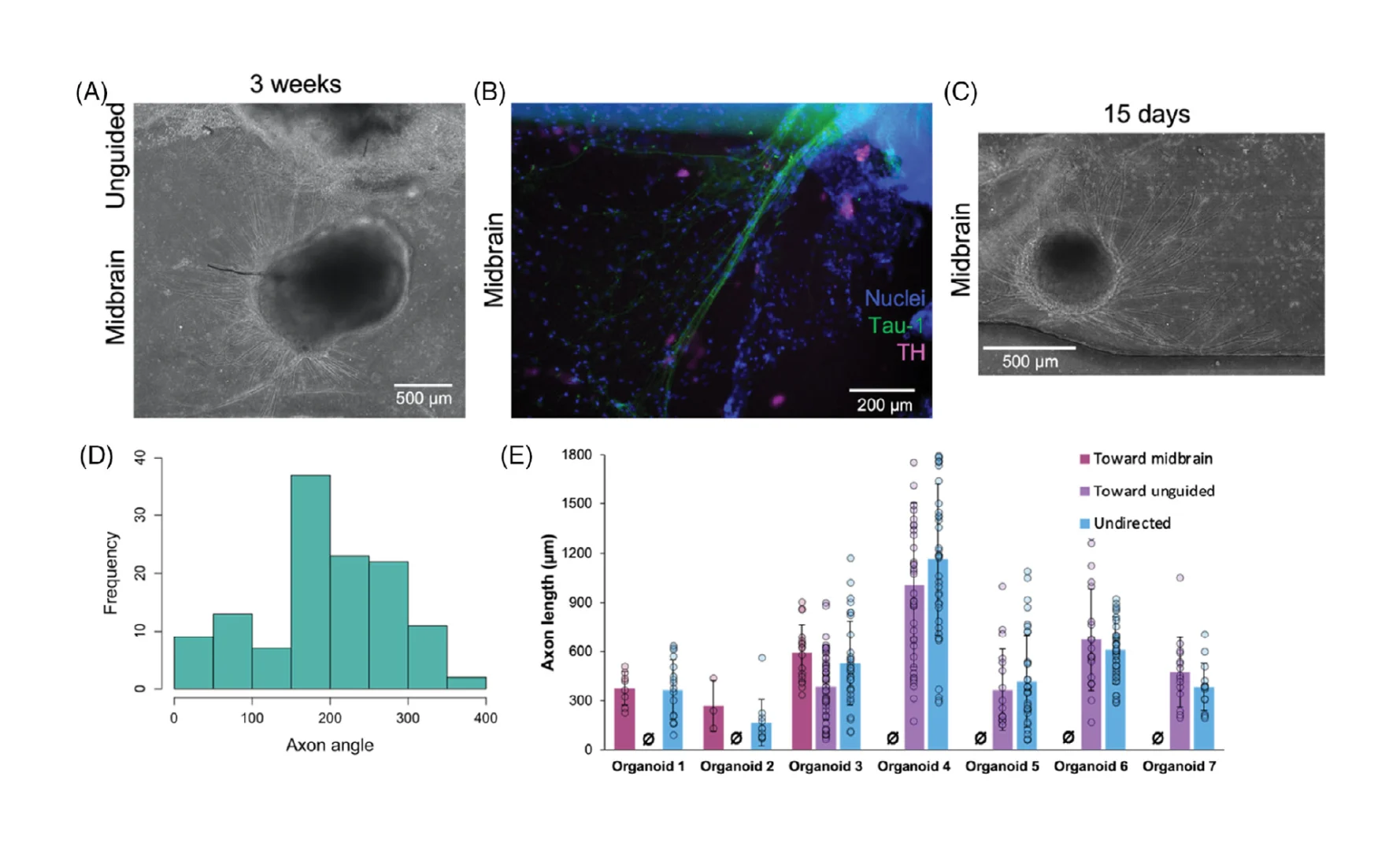
Based on mechanisms of axon guidance[17-19] it is reasonable to suppose that factors secreted during co-culture may direct axonal outgrowth. We first asked whether quantitative analysis would allow us to better understand the factors that might affect axon targeting behaviors. Axonal projections grew from all sides of the midbrain organoid (Figure 4C) toward midbrain organoids in the same channel (oriented at 180°), unguided organoids in the adjacent channel (oriented at 270°), and toward spaces without organoids in them. By comparing the frequency distributions of axonal direction (Figure 4D) against random chance, we found that axonal projections from midbrain organoids were significantly statistically biased toward other midbrain organoids (n = 50–159 axons from 3 to 4 organoids; p < 0.05 by one-sample t-test), while projections toward unguided organoids only approached significance (p < 0.1). In contrast, axon lengths were not significantly different regardless of direction toward which they grow (Figure 4E). This analysis would therefore suggest that when allowed to form in co-culture, the distribution of directed axon targeting may be biased by brain region-specific secreted factors.
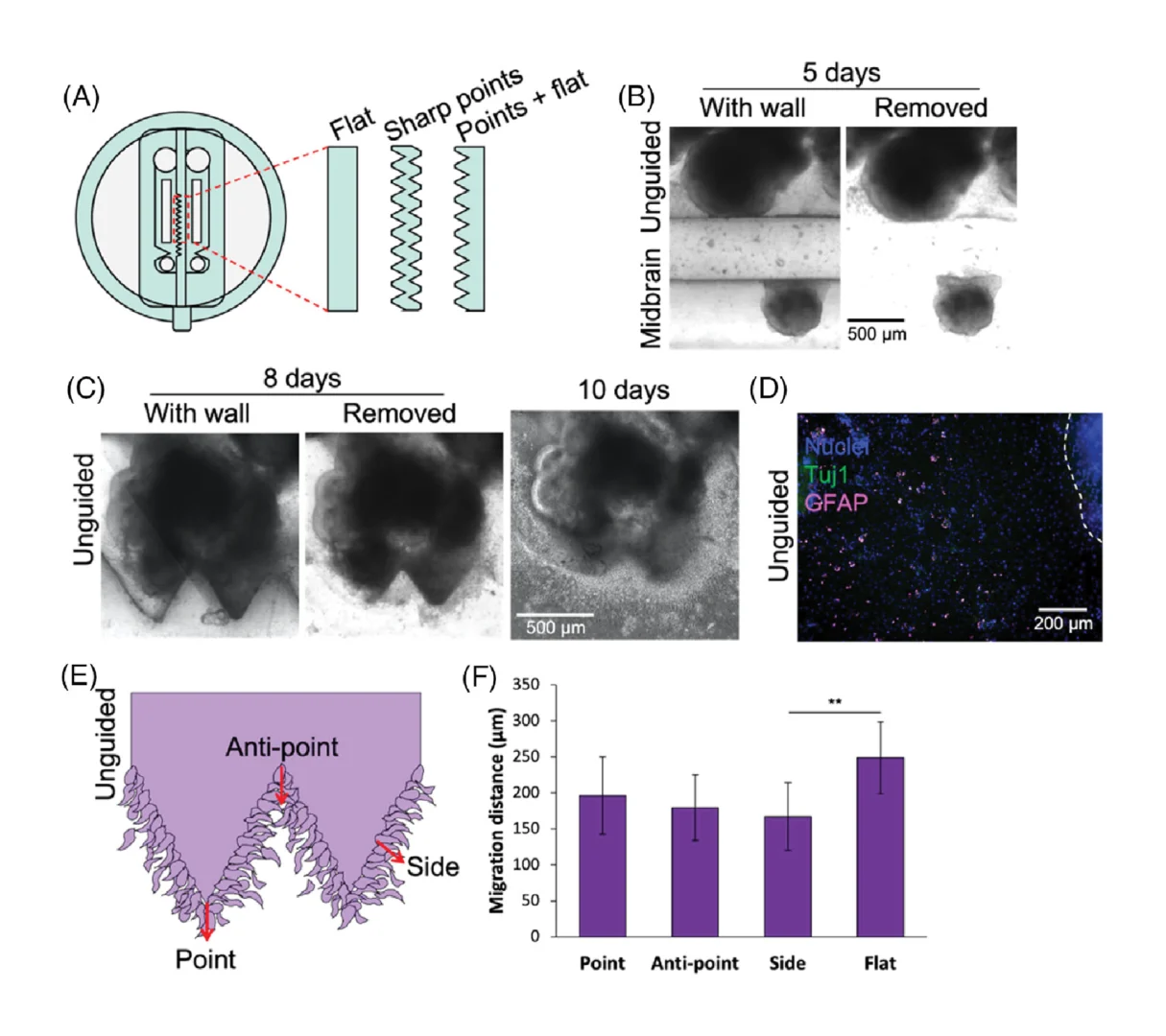
3.6. Organoid surface geometry influences cell migration
We also observed invasive migration of cells from the unguided organoids into the inter-organoidal space, and given the known impact of tissue geometry on cellular invasion and migration,[23-25] we asked whether organoid surface geometry might influence this invasive behavior during assembloid formation. We therefore tested flat versus triangular geometric shapes of the separating wall (Figure 5A), and the organoids grew to adopt the shapes provided by the channel wall (Figure 5B,C). Once the organoids had reached this stage, the separating inserts were removed. Interestingly, cells migrated out from the unguided organoids into the Matrigel towards the midbrain organoids regardless of organoid peripheral geometry (Figure 5C), and immunostaining indicated that some of these migrating cells were astrocytes, positive for GFAP (Figure 5D). However, migration distance was significantly different based on the global originating tissue shape (Figure 5E,F). Cells migrating from a flat organoid periphery travelled significantly farther than cells migrating from the flat midpoint of a triangle edge. Given that flat surfaces are predicted to have lower mechanical tension than surfaces of high curvature, it seems likely that differential mechanical priming arising from shape could lead to different migration distances.
4. Discussion
In this work, we build on recent advances by Park et al. in culturing shaped organoids[35] and develop a platform and methodology to individually culture multiple organoids of distinct types and control surface shape and spacing of the organoids, before allowing them to interact and form an assembloid on demand. Since the distance between shaped organoids can be precisely defined, the process of assembly can be closely observed in situ across multiple similarly-shaped live cultures during assembloid formation. Constructing devices capable of supporting long-term growth of organoids into predefined shapes, while affording the ability to (1) allow simultaneous but separate culture protocols for each of the organoid types, (2) support distinct surface modification strategies to enhance or prevent adhesion, and (3) to gently remove a separating barrier on demand required complex device geometries. To meet these fabrication demands, we developed a 3D printer-supported double-sided molding technique, which we successfully demonstrated to create barriers as thin as ≈200 µm. This fabrication method has the advantage of being extremely rapid and versatile, allowing design-to-device turnaround times of less than 8 h, while creating novel structures that would be extremely difficult to produce using conventional single-side replica molding approaches.
As a first demonstration of this technology, we investigated assembloid formation processes between midbrain and unguided organoids. Although we focused our experiments on controlling the space between organoids in separate channels, in principle this approach could in future be adapted to control the spacing between individual organoids, by reducing the length of the channel. Despite the simplicity of the current design, we were able to position organoids sufficiently close together to observe axonal projections from the midbrain organoids, and invasion of individual cells from the unguided organoids into the surrounding matrix. Together, these processes should capture key features of the assembloid formation process as two organoids merge with each other, which would be quite challenging to observe over time and quantitatively evaluate when simply placing optically-dense organoids against each other. Using the capacity for microfluidics to position and support these interactions, and to facilitate longitudinal observation using brightfield imaging, we were then able to demonstrate that axonal projections from the midbrain organoids display differential targeted outgrowth. Speculatively, since dopaminergic neurons project to a variety of brain regions in vivo,[51] these findings may ultimately be relevant in understanding how and why neuronal connections form differently in different regions of the brain. We were also able to observe that cell invasion from the unguided organoids was affected by global surface geometries. Unexpectedly, cells leaving flat organoids displayed higher invasive potential than cells leaving the flat regions of triangular shapes. This was unexpected because higher endogenous mechanical stress levels at shape-driven stress points such as triangular apexes have previously been thought of as “launching sites” for invasive cells in cancer models,[23-25] but the opposite was observed in our neural cultures. Taken together, these results suggest that the process of assembloid formation can be dissected using microfluidic systems, and that this general approach might be leveraged to improve our understanding of the development of neural circuitry, in healthy and diseased states, both within the brain and targeting of other organs such as muscle or gut. More generally, our observations that assembloid-formation behaviors can be directed through physical cues in the local microenvironment suggests that such approaches may ultimately be useful in establishing predictive control over complex assembloid formation processes.
Several limitations should be considered in the utility of these devices. First, although the experiments here were designed primarily to prove the concept of these devices using brain organoid components, applying this strategy to other organoids may present unexpected complications. For example, the device architecture was sufficient to support the metabolic needs for brain organoid maturation, but other more energy-intensive processes may require alternative designs or other strategies to enhance metabolic transport and availability. Second, the capacity to observe assembloid-associated processes between organoids is enhanced through our microfluidic system, but is ultimately limited by difficulties in imaging through optically-dense organoids. Strategies such as brain organoid tissue clearing for end-point analysis[52] and genetically-encoded markers to monitor processes in real time may be useful here, but cannot be used in parallel to allow for live, high-resolution imaging of such structures. Alternative imaging modalities such as MicroCT or ultrasound imaging may also have significant value in addressing this specific issue. Finally, the true capacity for assembloid formation to accurately capture in vivo processes and final structures remains unclear. While we hope that the devices presented here will provide important tools in answering such questions, much remains to be done in establishing the fundamental utility of assembloids as in vitro models of development and disease.